2次元コード¶
2次元コードはMPI並列版のみで、以下のような構成になっています。
Makefileは本2次元コードのバイナリ生成、削除をコントロールしています。初期化するには$PCANS_DIR/em2d_mpi内で、
$ make clean
としてください。Makefile_incには、
FC = mpif90
FFLAGS = -O2
が記述されており、makeする際の環境変数が設定されています。ここでは、"$FC"にはFortranコンパイラ、"$FFLAGS"にはコンパイラオプションが設定されています。コンパイラとコンパイラオプションを変更したい場合は、例えば、
FC = mpiifort
FFLAGS = -O3
のように修正してください。
物理課題として「 線形波動 (md_wave) 」、「 Weibel不安定 (md_weibel) 」、「 磁気リコネクション(md_mrx) 」、「 衝撃波(md_shock) 」、「 KH不安定(md_kh) 」が用意されています(2018年8月現在)。基本的な使い方は 1次元 と同じです。2次元MPI並列化コードによる違いを以下に示します。
領域分割法による並列化¶
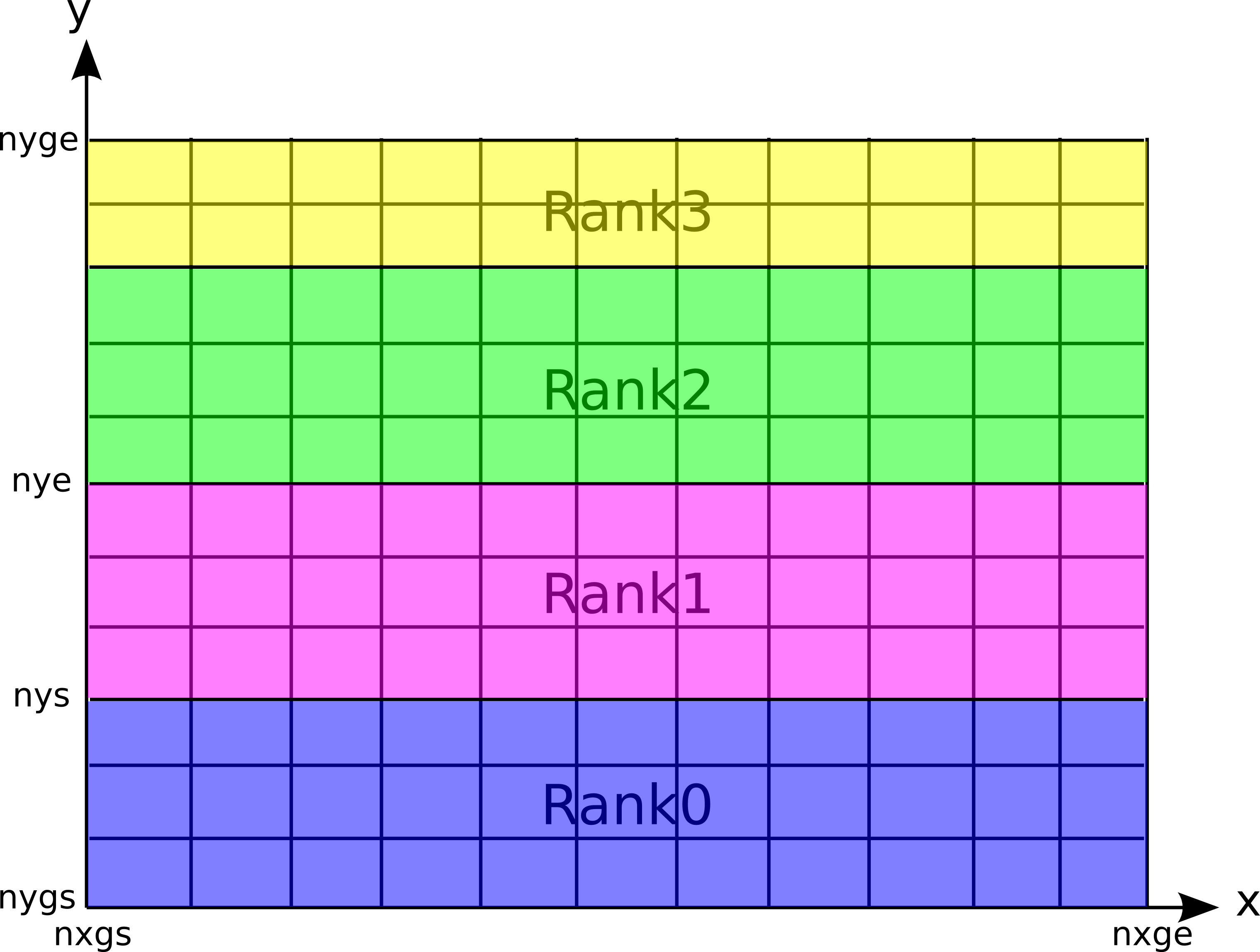
2次元コードにおける領域分割法と各プロセスが担当する領域¶
2次元コードでは、 y方向に1次元的に領域を区分化し、各領域にある粒子の運動と場の発展を各プロセス(Rank)が分担することにより、並列化を行っています( 2次元コードにおける領域分割の図 )。
注釈
領域分割による粒子コードの並列化では、系の発展に伴う粒子数の不均一化によって各プロセスの計算負荷が大きく異なることが考えられます。その場合、一番大きな負荷のプロセスによって計算時間が決まるため、並列化効率が落ちることが知られています(ロードバランスの非均衡化)。各物理課題では、y方向になるべく粒子数の偏りが少なくなるような初期設定をする必要があります(背景流れ場、磁場の向きなど)。
パラメタ設定¶
シミュレーション定数の設定(const.f90)は、以下のようになっています。
const.f90:
module const
implicit none
!!************************ NUMERICAL CONSTANTS ***********************************!!
integer, parameter :: nx = 128 ! NUMBER OF GRID POINTS IN X
integer, parameter :: ny = 128 ! NUMBER OF GRID POINTS IN Y
integer, parameter :: nxgs = 2 ! START POINT IN X
integer, parameter :: nxge = nxgs+nx-1 ! END POINT
integer, parameter :: nygs = 2 ! START POINT IN Y
integer, parameter :: nyge = nygs+ny-1 ! END POINT
integer, parameter :: np = 50*nx ! CUMULATIVE NUMBER OF PARTICLES IN EACH Y POSITION
integer, parameter :: nsfo = 1 ! SHAPE FUNCTION ORDER (0:NGP, 1:CIC, 2:SPLINE)
integer, parameter :: nsp = 2 ! NUMBER OF PARTICLE SPECIES
integer, parameter :: bc = 0 ! BOUNDARY CONDITION (PERIODIC:0, REFLECTIVE:-1)
integer, parameter :: nproc = 8 ! NUMBER OF PROCESSORS
!! SETUP FOR SUBROUTINES CALLED IN MAIN PROGRAM
integer, parameter :: itmax = 2048 !NUMBER OF ITERATION
integer :: it0 = 0 !0:INITIAL, NONZERO/9999999: RESTART DATA
integer, parameter :: intvl1 = 10 !INTERVAL FOR PARTICLES & FIELDS STORAGE
integer, parameter :: intvl2 = 100 !INTERVAL FOR ENERGY CALC.
character(len=128) :: dir = './dat/' !DIRECTORY FOR OUTPUT
character(len=128) :: fname_param = 'init_param.dat' !FILE NAME OF INIT CONDITIONS
character(len=128) :: fname_energy = 'energy.dat' !FILE NAME OF PARTICLE DATA
!! OTHER CONSTANTS
real(8), parameter :: gfac = 0.501D0 !IMPLICITNESS FACTOR > 0.5
real(8), parameter :: cfl = 1.0D0 !CFL CONDITION FOR LIGHT WAVE
real(8), parameter :: delx = 1.0D0 !CELL WIDTH
real(8), parameter :: rdbl = 1.0D0 !DEBYE LENGTH / CELL WIDTH
real(8), parameter :: pi = 4.0D0*atan(1.0D0)
!!************************ PHYSICAL CONSTANTS ***********************************!!
!! n0 : NUMBER OF PARTICLES/CELL
!! c : SPEED OF LIGHT
!! mr : ION-TO-ELECTRON MASS RATIO
!! alpha : wpe/wge = c/vth_e * sqrt(beta_e)
!! beta : ION PLASMA BETA
!! rtemp : Te/Ti
integer, parameter :: n0 = 25
real(8), parameter :: c = 1.0D0
real(8), parameter :: mr = 16.0D0
real(8), parameter :: alpha = 2.0D0, beta = 0.1D0, rtemp=1.0D0
end module
MPI並列版では、"nxgs"、"nxge"、"nygs"、"nyge"が設定されていて、グリッド番号の最初の値(nxgs、nygs)を任意に指定しています。"nproc"はプロセス数で、MPIで並列化するプロセス数を指定します(この例では8並列)。
本コードでは、2次元領域分割は行わず、y方向に1次元領域分割をしています( 2次元コードにおける領域分割の図 )。各担当領域を表す"nys"、"nye"(上図参照)は、init.f90内で、"nygs"、"nyge"、"nproc"から求めています。また、y方向は周期境界条件を仮定しています(x方向はbcで指定可能)。このような制限があるものの、多くの2次元物理問題に対して適用可能です(衝撃波、Weibel不安定、磁気リコネクション、KH不安定など)。また、2次元コードでは粒子の形状関数を選ぶことができるようになっています。nsfoを0 (NGP)、1 (CIC)、2 (spline)に設定することにより選ぶことができます。デフォルトはCIC(nsfo=1)です。
粒子データの解析¶
1次元シリアル版 では、モーメントを計算すると同時に粒子データを各課題ディレクトリ内の"psd/"内に出力していましたが、2次元計算では粒子データは膨大なものになることが予想されるため、粒子データの出力のために別途プログラムが用意されています("$PCANS_DIR/em2d_mpi/psd/"内)。以下ではその使い方について説明します。
"$PCANS_DIR/psd/"内には、指定した位置(x,y)の回り(±2グリッド)にある粒子を選んで、粒子の位置・速度を出力するプログラムが格納されています。各課題で計算が終わったのち、
$ make psd_calc
としてください。"dat/"内にある粒子と電磁場データを読み込み、計算結果を"psd/"内に出力します。Makefile内の該当個所は、
############## psd calculation ################
psd_calc: $(LIB_DIR)/psd/psd.out
$(LIB_DIR)/psd/psd.out 100 100 ./ ./dat/*_rank*.dat
$(LIB_DIR)/psd/psd.out :
cd $(LIB_DIR)/psd ; make
###############################################
です。引数として注目するグリッド位置(この例の場合は 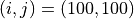 )を与えています。解析する際はこの値を変更してください。デフォルトでは"dat/"内にある粒子データを全て計算することになりますが、"./dat/*_rank*.dat"を修正することにより、任意の時間ステップにおける粒子データに対して処理を行います。
)を与えています。解析する際はこの値を変更してください。デフォルトでは"dat/"内にある粒子データを全て計算することになりますが、"./dat/*_rank*.dat"を修正することにより、任意の時間ステップにおける粒子データに対して処理を行います。
